

Contributions
Abstract: LB2604
Type: Oral Presentation
Presentation during EHA23: On Sunday, June 17, 2018 from 12:00 - 12:15
Location: Room A1
Background
SETBP1 variants occur as somatic mutations in several hematological malignancies such as atypical Chronic Myeloid Leukemia and as de novo germline mutations in the Schinzel-Giedion Syndrome (SGS), a disorder characterized by severe intellectual disability and multi-organ development abnormalities.
Aims
The aim of this work is to dissect the role of SETBP1 as an epigenetic modulator of gene transcription.
Methods
ChIP-Seq, RNA-Seq, Co-Immunoprecipitation, HPLC-Mass Spectrometry, ATAC-Seq, in-utero electroporation.
Results
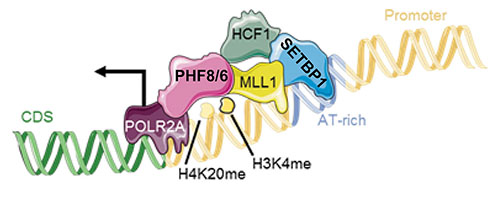
ChIP-Seq experiments performed on 293 FLP-In lines harboring WT and mutated (G870S) SETBP1 revealed 3065 broad genomic regions bound by SETBP1-G870S. The peaks clustered in AT-rich regions (p<0.0001); analysis of these regions using de novo motif discovery tools led to the identification of a SETBP1 consensus binding site (AAAATAA/T; p=0.002). Notably, this consensus largely overlapped the classical AT-hook motif (AAAATA), suggesting that SETBP1 may bind gDNA through its AT-hook domains.
RNA-Seq analyses performed on G870S as well as Empty control lines identified 2687 differentially expressed genes (DEGs; 57% down- and 43% up-regulated). The intersection between genes bound by SETBP1 in promoter regions and DEGs (FDR<0.001) revealed 105 co-occurring genes. The vast majority of these genes was upregulated (99; 94.3%; p<1x10-6), suggesting a role for SETBP1 as a positive inducer of gene expression. Co-immunoprecipitation/proteomics and FRET experiments showed a direct interaction between SETBP1 and HCF1, a core protein of the KMT2A (MLL1) complex, mainly responsible for H3K4 mono- and di-methylation.
In-silico linear domain analysis suggested the presence of a HCF1 binding motif (HBM) occurring at position 991-994 of SETBP1. Notably, HCF1 is known to be part of the KMT2A core complex. Deletion of this region of SETBP1 caused the abrogation of the SETBP1/HCF1 interaction and complete normalization of the expression level of SETBP1 target genes, therefore suggesting that SETBP1 and KMT2A are part of the same complex. Co-IP experiments confirmed this hypothesis (Fig. 1).
ChIP-Seq data also showed that SETBP1 binds to the MECOM oncogene promoter. In line with these findings, MECOM and MECOM target genes were differentially expressed both in cells expressing SETBP1-G870S (p<0.0001) and in 32 aCML cases (11 positive and 21 negative for SETBP1 somatic mutations), where SETBP1-positive patients expressed higher levels of MECOM (p=0.0002) and MECOM target genes (p<0.0001).
Transduction of radial glial progenitors of cerebral cortices of E13.5 mouse embryos with an expression plasmid encoding SETBP1-G870S using an in utero electroporation system caused an impairment of mouse embryo neurogenesis with a profound delay in neuronal migration, indicating that SETBP1 is directly responsible for the neuroanatomical defects described in SGS.
Conclusion
In summary, this work unveils for the first time a new SETBP1 function which directly promotes gene transcription and clarifies a new mechanism operating in myeloid malignancies and in SGS.
Session topic: 15. Myeloproliferative neoplasms – Biology & Translational Research
Keyword(s): MLL, Epigenetic, Transcriptional regulation, myeloid leukemia
Abstract: LB2604
Type: Oral Presentation
Presentation during EHA23: On Sunday, June 17, 2018 from 12:00 - 12:15
Location: Room A1
Background
SETBP1 variants occur as somatic mutations in several hematological malignancies such as atypical Chronic Myeloid Leukemia and as de novo germline mutations in the Schinzel-Giedion Syndrome (SGS), a disorder characterized by severe intellectual disability and multi-organ development abnormalities.
Aims
The aim of this work is to dissect the role of SETBP1 as an epigenetic modulator of gene transcription.
Methods
ChIP-Seq, RNA-Seq, Co-Immunoprecipitation, HPLC-Mass Spectrometry, ATAC-Seq, in-utero electroporation.
Results
ChIP-Seq experiments performed on 293 FLP-In lines harboring WT and mutated (G870S) SETBP1 revealed 3065 broad genomic regions bound by SETBP1-G870S. The peaks clustered in AT-rich regions (p<0.0001); analysis of these regions using de novo motif discovery tools led to the identification of a SETBP1 consensus binding site (AAAATAA/T; p=0.002). Notably, this consensus largely overlapped the classical AT-hook motif (AAAATA), suggesting that SETBP1 may bind gDNA through its AT-hook domains.
RNA-Seq analyses performed on G870S as well as Empty control lines identified 2687 differentially expressed genes (DEGs; 57% down- and 43% up-regulated). The intersection between genes bound by SETBP1 in promoter regions and DEGs (FDR<0.001) revealed 105 co-occurring genes. The vast majority of these genes was upregulated (99; 94.3%; p<1x10-6), suggesting a role for SETBP1 as a positive inducer of gene expression. Co-immunoprecipitation/proteomics and FRET experiments showed a direct interaction between SETBP1 and HCF1, a core protein of the KMT2A (MLL1) complex, mainly responsible for H3K4 mono- and di-methylation.
In-silico linear domain analysis suggested the presence of a HCF1 binding motif (HBM) occurring at position 991-994 of SETBP1. Notably, HCF1 is known to be part of the KMT2A core complex. Deletion of this region of SETBP1 caused the abrogation of the SETBP1/HCF1 interaction and complete normalization of the expression level of SETBP1 target genes, therefore suggesting that SETBP1 and KMT2A are part of the same complex. Co-IP experiments confirmed this hypothesis (Fig. 1).
ChIP-Seq data also showed that SETBP1 binds to the MECOM oncogene promoter. In line with these findings, MECOM and MECOM target genes were differentially expressed both in cells expressing SETBP1-G870S (p<0.0001) and in 32 aCML cases (11 positive and 21 negative for SETBP1 somatic mutations), where SETBP1-positive patients expressed higher levels of MECOM (p=0.0002) and MECOM target genes (p<0.0001).
Transduction of radial glial progenitors of cerebral cortices of E13.5 mouse embryos with an expression plasmid encoding SETBP1-G870S using an in utero electroporation system caused an impairment of mouse embryo neurogenesis with a profound delay in neuronal migration, indicating that SETBP1 is directly responsible for the neuroanatomical defects described in SGS.
Conclusion
In summary, this work unveils for the first time a new SETBP1 function which directly promotes gene transcription and clarifies a new mechanism operating in myeloid malignancies and in SGS.
Session topic: 15. Myeloproliferative neoplasms – Biology & Translational Research
Keyword(s): MLL, Epigenetic, Transcriptional regulation, myeloid leukemia