
THALASSEMIA IN MADRID: A PICTURE OF THE CURRENT SITUATION
(Abstract release date: 05/18/17)
EHA Library. J. Bardón-Cancho E. 05/18/17; 182913; PB2200

Eduardo J. Bardón-Cancho
Contributions
Contributions
Abstract
Abstract: PB2200
Type: Publication Only
Background
Diagnosis of thalassemia (Thal) in a Mediterranean country as Spain, could be thought as endemic, but few data are available so far. Moreover, attention to hemoglobinopathies is focused on sickle cell disease.
Aims
The aim of our study was to find out the prevalence of Thal and clinical significant hemoglobinopathies other than sickle cell diseases in a referral center for newborn sickle screening, in addition to the demographic characteristics of these patients. The secondary objectives were to obtain the frequency of specific treatments or prophylaxis accomplished by these patients, and the reasons for loss to follow-up.
Methods
The study is observational, unicentric, descriptive and retrospective, carried out in December 2016 in a tertiary hospital in the Community of Madrid, Spain. All patients diagnosed with Thal and other not sickle-hemoglobinopathies who had attended at least once to the hematology clinic were included. Demographic characteristics (date of birth, gender, country of birth) and clinical ones (genotype or Thal type, therapy and update in follow up, like alive, deceased or lost patient) were collected. Written informed consent was signed by patients or legal guardians in accordance with the Declaration of Helsinki. The study was approved by the hospital Ethical Committee. Statistical analyses were performed using SPSS version 18.0. Quantitative variables were reported as median or mean value and range, while categorical variables were expressed as absolute value and percentage.
Results
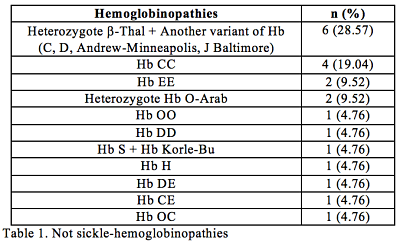
The total number of patients included was 31 (9 Thalassemia Major (TM), 1 Thalassemia Intermedia (TI), 21 other not sickle-hemoglobinopathies). The center follows 209 sickle patients, which leads to a ratio sickle/not sickle of 6.74 (Table 1). Ratio boy/girl is 1.21 for all group. Most of patients were born in Spain (90.32 %), although 6.45 % were born in Asia and one patient was born in Rumania. Considering the parents, 32% were born in Europe, 29% from Africa, 25% from Asia, and 12% from America. 92% of those patients born in Spain were detected in their first days of life due to universal screening detection implemented in Madrid since 2003. Median age at first diagnosis was 0.70 years (0-16.35). Median age at the end of inclusion was 9.39 years (range 1.90 to 35.44). 35% of them had molecular genotyping for diagnostic confirmation. Two out of 10 patients with Thal had HLA identical siblings.
Quelation treatment was added to standard treatment to all the patients with Thal: 7 received deferasirox, 3 were treated with deferoxamine and 2 with deferiprone; 2 of the patients required double quelation. Two out of 10 patients with Thal underwent splenectomy. None of these patients had sepsis or meningitis.
Three Thal patients underwent progenitor stem cell transplantations and they remain on complete chimerism in the present moment.
Patients lost to follow-up summed up 14: 3 emigrated to other countries, 2 continue the monitor of their diseases in other centers or in adults units and 7 for unknown reasons. There was one death (3.22 %) for a cause unrelated to his illness.
Conclusion
Early diagnosis derived from universal neonatal screening for sickle cell disease allows an effective health education and prompt therapy to other hemoglobinopathies, and a correct and thorough follow-up of these patients.
Session topic: 26. Thalassemias
Keyword(s): Hemoglobin variants, epidemiology, Diagnosis, Thalassemia
Abstract: PB2200
Type: Publication Only
Background
Diagnosis of thalassemia (Thal) in a Mediterranean country as Spain, could be thought as endemic, but few data are available so far. Moreover, attention to hemoglobinopathies is focused on sickle cell disease.
Aims
The aim of our study was to find out the prevalence of Thal and clinical significant hemoglobinopathies other than sickle cell diseases in a referral center for newborn sickle screening, in addition to the demographic characteristics of these patients. The secondary objectives were to obtain the frequency of specific treatments or prophylaxis accomplished by these patients, and the reasons for loss to follow-up.
Methods
The study is observational, unicentric, descriptive and retrospective, carried out in December 2016 in a tertiary hospital in the Community of Madrid, Spain. All patients diagnosed with Thal and other not sickle-hemoglobinopathies who had attended at least once to the hematology clinic were included. Demographic characteristics (date of birth, gender, country of birth) and clinical ones (genotype or Thal type, therapy and update in follow up, like alive, deceased or lost patient) were collected. Written informed consent was signed by patients or legal guardians in accordance with the Declaration of Helsinki. The study was approved by the hospital Ethical Committee. Statistical analyses were performed using SPSS version 18.0. Quantitative variables were reported as median or mean value and range, while categorical variables were expressed as absolute value and percentage.
Results
The total number of patients included was 31 (9 Thalassemia Major (TM), 1 Thalassemia Intermedia (TI), 21 other not sickle-hemoglobinopathies). The center follows 209 sickle patients, which leads to a ratio sickle/not sickle of 6.74 (Table 1). Ratio boy/girl is 1.21 for all group. Most of patients were born in Spain (90.32 %), although 6.45 % were born in Asia and one patient was born in Rumania. Considering the parents, 32% were born in Europe, 29% from Africa, 25% from Asia, and 12% from America. 92% of those patients born in Spain were detected in their first days of life due to universal screening detection implemented in Madrid since 2003. Median age at first diagnosis was 0.70 years (0-16.35). Median age at the end of inclusion was 9.39 years (range 1.90 to 35.44). 35% of them had molecular genotyping for diagnostic confirmation. Two out of 10 patients with Thal had HLA identical siblings.
Quelation treatment was added to standard treatment to all the patients with Thal: 7 received deferasirox, 3 were treated with deferoxamine and 2 with deferiprone; 2 of the patients required double quelation. Two out of 10 patients with Thal underwent splenectomy. None of these patients had sepsis or meningitis.
Three Thal patients underwent progenitor stem cell transplantations and they remain on complete chimerism in the present moment.
Patients lost to follow-up summed up 14: 3 emigrated to other countries, 2 continue the monitor of their diseases in other centers or in adults units and 7 for unknown reasons. There was one death (3.22 %) for a cause unrelated to his illness.
Conclusion
Early diagnosis derived from universal neonatal screening for sickle cell disease allows an effective health education and prompt therapy to other hemoglobinopathies, and a correct and thorough follow-up of these patients.
Session topic: 26. Thalassemias
Keyword(s): Hemoglobin variants, epidemiology, Diagnosis, Thalassemia
{{ help_message }}
{{filter}}