

Contributions
Abstract: S439
Type: Oral Presentation
Presentation during EHA22: On Saturday, June 24, 2017 from 12:15 - 12:30
Location: Room N105
Background
Aims
Investigating the pathogenic role of the recurrent R98S mutation in ribosomal protein L10 (RPL10) in T-ALL.
Methods
Results
The differential proteome screen revealed overexpression of several Jak-Stat signaling components (Csf2rb/2, Jak1, Stat1, Stat3, Stat5a/b and Stat6) in engineered RPL10 R98S mouse lymphoid cells, which we confirmed in hematopoietic cells derived from a transgenic RPL10 R98S mouse model. The relevance of this overexpression was illustrated by enhanced Jak-Stat pathway activation upon cytokine stimulation in RPL10 R98S lymphoid cells, as well as increased sensitivity of these cells to clinically used JAK-STAT inhibitors ruxolitinib and pimozide. RPL10 R98S positive leukemia patients likewise showed overexpression of IL7RA, JAK1 and STAT5, increased sensitivity to pimozide, as well as a mutually exclusive mutation pattern between RPL10 R98S and JAK-STAT lesions, suggesting that RPL10-R98S also modulates the cascade in human T-ALL. Programmed -1 ribosomal frameshifting (-1 PRF) recently emerged as a post-transcriptional mechanism regulating expression of cytokine receptors. We identified -1 PRF signals in mouse and human Jak-Stat genes and observed RPL10 R98S associated frameshifting reduction in several of these, which may contribute to their overexpression. Altered levels of -1 PRF can however only partially explain observed JAK-STAT protein expression changes, and transcriptional changes and altered protein stability are also involved. Indeed, our data point to altered proteasome activity and composition in RPL10 R98S cells, with upregulation of immunoproteasome specific catalytic subunits, which may explain the increased stability of particular proteins such as Jak1. Of further medical interest, RPL10 R98S cells showed reduced proteasome activity and enhanced sensitivity to the clinically used proteasome inhibitors bortezomib and carfilzomib.
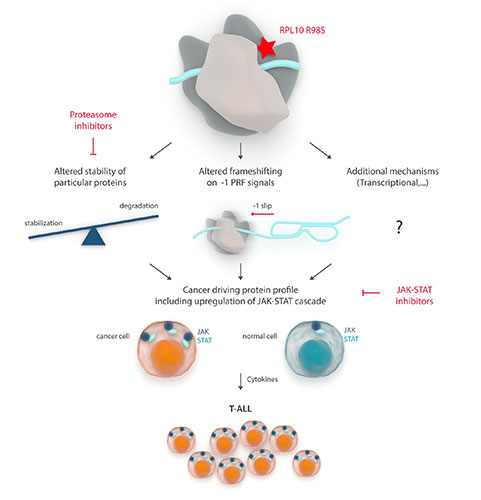
Conclusion
Session topic: 1. Acute lymphoblastic leukemia - Biology
Abstract: S439
Type: Oral Presentation
Presentation during EHA22: On Saturday, June 24, 2017 from 12:15 - 12:30
Location: Room N105
Background
Aims
Investigating the pathogenic role of the recurrent R98S mutation in ribosomal protein L10 (RPL10) in T-ALL.
Methods
Results
The differential proteome screen revealed overexpression of several Jak-Stat signaling components (Csf2rb/2, Jak1, Stat1, Stat3, Stat5a/b and Stat6) in engineered RPL10 R98S mouse lymphoid cells, which we confirmed in hematopoietic cells derived from a transgenic RPL10 R98S mouse model. The relevance of this overexpression was illustrated by enhanced Jak-Stat pathway activation upon cytokine stimulation in RPL10 R98S lymphoid cells, as well as increased sensitivity of these cells to clinically used JAK-STAT inhibitors ruxolitinib and pimozide. RPL10 R98S positive leukemia patients likewise showed overexpression of IL7RA, JAK1 and STAT5, increased sensitivity to pimozide, as well as a mutually exclusive mutation pattern between RPL10 R98S and JAK-STAT lesions, suggesting that RPL10-R98S also modulates the cascade in human T-ALL. Programmed -1 ribosomal frameshifting (-1 PRF) recently emerged as a post-transcriptional mechanism regulating expression of cytokine receptors. We identified -1 PRF signals in mouse and human Jak-Stat genes and observed RPL10 R98S associated frameshifting reduction in several of these, which may contribute to their overexpression. Altered levels of -1 PRF can however only partially explain observed JAK-STAT protein expression changes, and transcriptional changes and altered protein stability are also involved. Indeed, our data point to altered proteasome activity and composition in RPL10 R98S cells, with upregulation of immunoproteasome specific catalytic subunits, which may explain the increased stability of particular proteins such as Jak1. Of further medical interest, RPL10 R98S cells showed reduced proteasome activity and enhanced sensitivity to the clinically used proteasome inhibitors bortezomib and carfilzomib.
Conclusion
Session topic: 1. Acute lymphoblastic leukemia - Biology