Abstract: S430
Type: Oral Presentation
Presentation during EHA22: On Saturday, June 24, 2017 from 12:30 - 12:45
Location: Hall E
Background
Acute myeloid leukaemia (AML) patient survival remains below 30% and there have been no major new anti-AML therapies for decades.
Aims
To identify novel therapeutic vulnerabilities of AML
Methods
A CRISPR-Cas9 drop-out screen was used to identify genetic vulnerabilities of AML. Downstream, we study the function of the RNA methyltransferase METTL3, a novel therapeutic vulnerability of AML. These included in-vitro and in-vivo validation of METTL3 as a therapeutic target using CRISPR/gRNA or shRNA, and investigation of its function using ChIP-seq, RNA-IP-seq, ribosome footprinting (RFP) and bioinformatic analyses.
Results
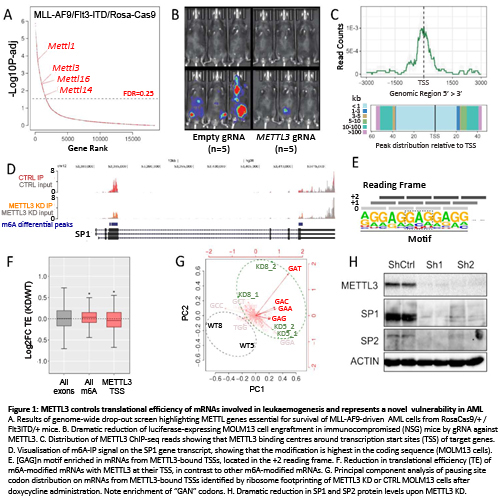
We performed a genome-wide CRISPR screen on AML cells from Flt3ITD/+;RosaCas9/+ mice transformed with MLL-AF9 lentivirus and identified >1500 cell-essential genes of which ~250 were AML-specific and included many MLL-AF9 interactors and several putative RNA methyltransferase genes: METTL1, METTL3, METTL14 and METTL16 (Fig 1A). Focusing on METTL3, we show that its disruption with Cas9/gRNA promoted differentiation of murine and human MLL-AF9 AML cells and inhibited their growth in vitro and in vivo (Fig 1B), but did not affect primary murine haematopoietic stem/progenitor cells.
To investigate METTL3 function we performed chromatin immunoprecipitation (ChIP) for METTL3 and H3K4me3 and identified 126 METTL3 peaks, localized mainly at promoters with bimodal H3K4me3. METTL3 binding was highest at transcription start sites (TSS) (Fig 1C) and the most enriched transcription factor motif at METTL3 sites was that for the NFY complex. Using available ChIP-seq datasets we found that NFYA, NFYB, H3R2me2s, WDR5 and KLF9 showed strong co-binding with METTL3. Also shRNA knock-down (KD) of WDR5, led to reduced METTL3 binding to target genes SP1 and SP2.
To investigate if/how METTL3 controls expression of target genes we first noted that their mRNA levels were unaffected by METTL3 KD. As METTL3 is an N6-methyladenosine (m6A) methyltranferase, we then performed RNAseq after IP with an m6A-specific antibody (m6A-IP). This identified >4000 METTL3-dependent m6A peaks on poly-A+ RNA. m6A peaks were seen on 72.4% of METTL3-bound gene transcripts and were located in the coding region (CDS) in contrast to STOP codon enrichment in the general transcriptome (Fig 1D). Also, METTL3-bound transcripts were enriched for the [GAG]n motif, which was almost always in the +2 reading frame (Fig1E). As this raised the possibility that m6A may regulate translation of these transcripts, we performed RFP on wild type (WT) and METTL3 KD MOLM13 cells, to evaluate translational efficiency (TE). Strikingly, upon METTL3 KD, m6A-marked genes had increased TE, whilst transcripts with METTL3 at their TSS had reduced TE (Fig1F). We then mapped ribosomal pausing sites on mRNAs from METTL3-bound TSSs and found that GAN codons were more occupied (paused) in METTL3 KD vs WT cells (Fig1G). To understand consequences at the protein level, we studied METTL3 targets SP1 and SP2. Remarkably, in contrast to their mRNA levels, SP1 and SP2 proteins were markedly reduced upon METTL3 KD (Fig1H). SP1 and SP2 targets including c-MYC, were also downregulated upon METTL3 KD and simultaneous SP1/SP2 gRNA KD markedly reduced proliferation of MLL-AF9/Flt3ITD/+ cells.
Conclusion
Our results show that METTL3 controls translation of specific mRNAs by binding their TSS and introducing m6A at [GAG]n motifs within their CDS, in turn increasing their TE. These mRNAs code for proteins essential for AML cell survival, making METTL3 a novel therapeutic vulnerability of AML.
Session topic: 3. Acute myeloid leukemia - Biology

